
Community Resources and Collaborations for Invertebrate Genomics Research
Welcome to newly elected 2024 GIGA board members:
President: Carlos Prada (University of Rhode Island)
Executive Vice President of Conferences: Jean-Francois Flot (Université Libre de Bruxelles)
Executive Vice President of Scholarships/Early Developments: Sadye Paez (The Rockefeller University)
Executive Vice President of External Development/Fund Raising: Joe Lopez (Nova Southeastern University)
Graduate Student/Postdoc representatives: Lisa Mesrop (University of California, Santa Barbara), João Gabriel R. N. Ferreira (Universidade Federal do Rio de Janeiro)
GIGA Data Repositories
- GIGA Zenodo Repository: Datasets and open-source publications
- GIGA NCBI BioProject: “Aquatic non-vertebrate metazoa”
Contribute to the GIGA NCBI BioProject and GIGA-GoaT projects
- Link your pre-existing BioProject to GIGA’s umbrella project.
- Add your genome projects to the GIGA-GoaT (Genomes on a Tree) species target lists.
- Utilize the GoaT resources for project planning and collaborations.
- Register with GIGA-NCBI BioProject and utilize the GIGA-GoaT Resources. 2023 GIGA-V slide deck (Dr. Jeffrey Robinson, UMBC).
Huge thanks to our individual donors and organizational sponsors:


Jack Krebs
Maribeth Glorioso
Shayma Zaidan
Lissa Messing
Emily Schmitt
Erlinda Dumas
GIGA Newsletter:
Liria Huberv
Andre Weisbrod
Kirk Dotson
Juan Armando Sánchez
GIGA V (2023) Conference
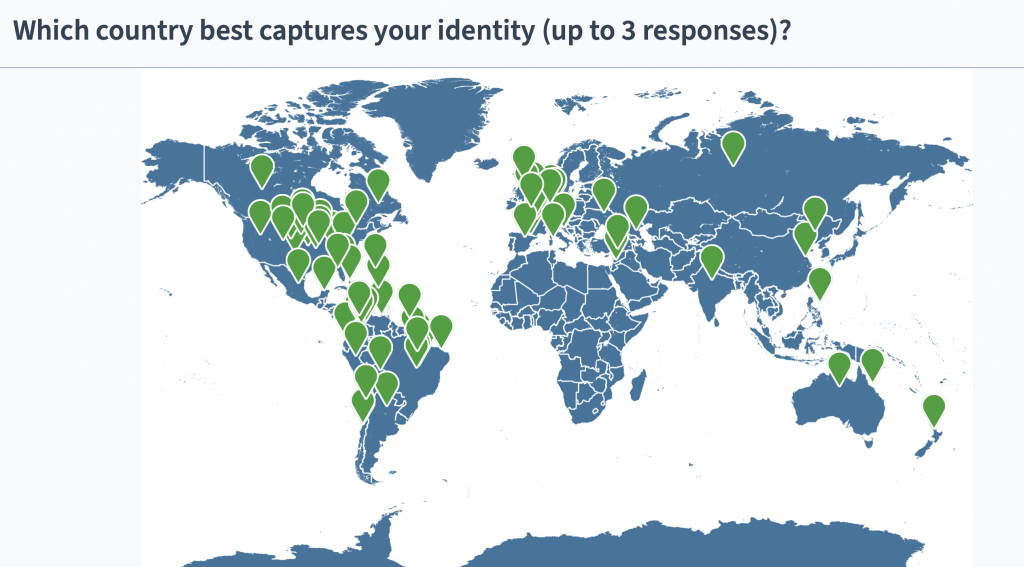
A snapshot of the GIGA Community:
-
Two New Sponge Genomes
New genome papers are out for Glass Sponges (Hexactinellida)!! This group of sponges is known for their unique silicate skeletal elements and deep-sea reefbuilding activities. Oopsacas minuta has a genome surprisingly devoid of metazoan core genes, while Aphrocallistes vastus is an important deep-sea reefbuilding species. Oopsacas minuta Santini S, Schenkelaars Q, Jourda C, Duchesne M,…
Latest News:
- Two New Sponge Genomes
- GIGA V conference, Cartagena, Colombia REGISTRATIONS OPEN
- GIGA officially a 501(c)(3) non-profit.
- GIGA fellowship recipient Kate Castellano on her research and career
- GIGA Board Elections Results
Donate and support the GIGA mission:
Funds are currently being allocated to education and training in genomics through graduate fellowship awards.
Donations can be submitted at https://www.paypal.com/paypalme2/GIGAIII.

Featured photography from GIGA collaborators:











GIGA III Conference and Workshop, October 2018
